



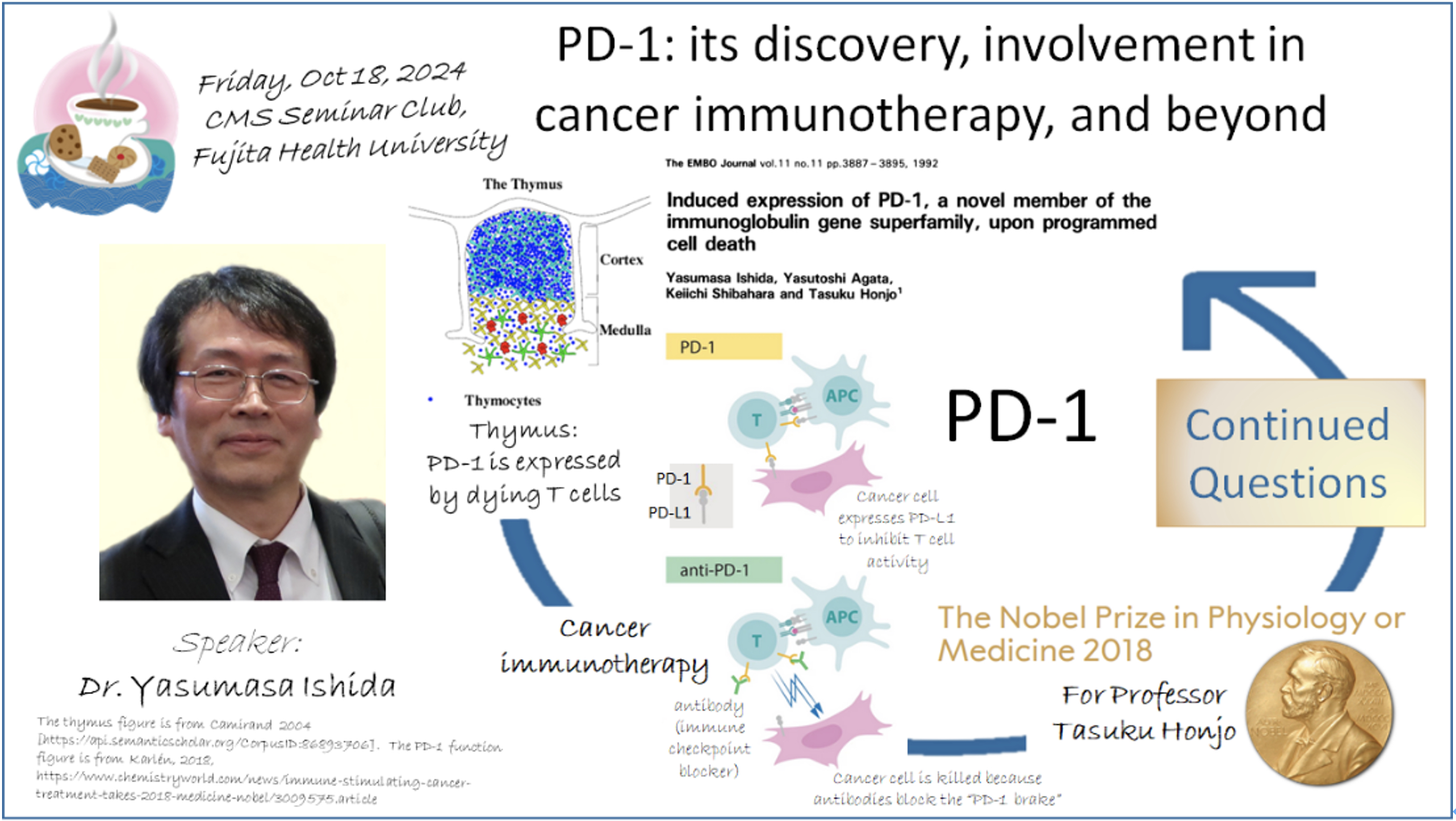
最近のがん治療の研究により、活性化されたTリンパ球(T細胞)上に発現が誘導されるPD-1分子の機能を抗体で阻害すると、がん細胞中のゲノム変異に起因するneoantigens への免疫応答が有意に回復することが分かりました。このことは、裏を返せば、がん患者さんの体内では、PD-1によってゲノム変異由来抗原への免疫応答が強く抑制されていることを意味します。では、一体なぜ、PD-1はがん細胞に対する特異的な免疫応答を抑制しなければならないのでしょうか?この疑問に答えるために、私たちは分子遺伝学と免疫学を架橋する領域において、挑戦的でユニークな研究を展開しています。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}